- Clark Center W350B
- cimprich@stanford.edu
Genomic instability contributes to many diseases, such as cancer, neurodegenerative disease, and developmental disorders, but it also underlies many natural processes including aging, evolution, and antibody diversification. The Cimprich lab is focused on understanding the mechanisms that the cell uses to maintain genomic stability, with an emphasis on the DNA damage response. This is a complex, multi-faceted response that requires cells to sense the presence of DNA damage within the vast genome, as well as to “choose” and coordinate a wide range of downstream events and outcomes. These include effects on DNA repair, transcription, and DNA replication, as well as arrest of cell cycle progression, apoptosis, and senescence.
We are particularly interested in understanding how DNA damage is identified and resolved during DNA replication. The genome is particularly vulnerable during DNA replication because the replisome stalls at many DNA lesions. Stalled replication forks are unstable and can be processed in aberrant ways, leading to double-strand break formation and chromosomal rearrangements. Thus, cells must rapidly stabilize stalled forks and initiate a pathway to restart and complete DNA replication.
The lab studies the DNA damage response using cultured mammalian cells, as well as cell-free extracts derived from the eggs of the frog Xenopus laevis. We use these systems and a range of multidisciplinary techniques to understand how the DNA damage response is initiated, how this pathway is integrated with the processes of DNA replication and transcription, and how cells recover from DNA damage. Specific areas of current interest are:
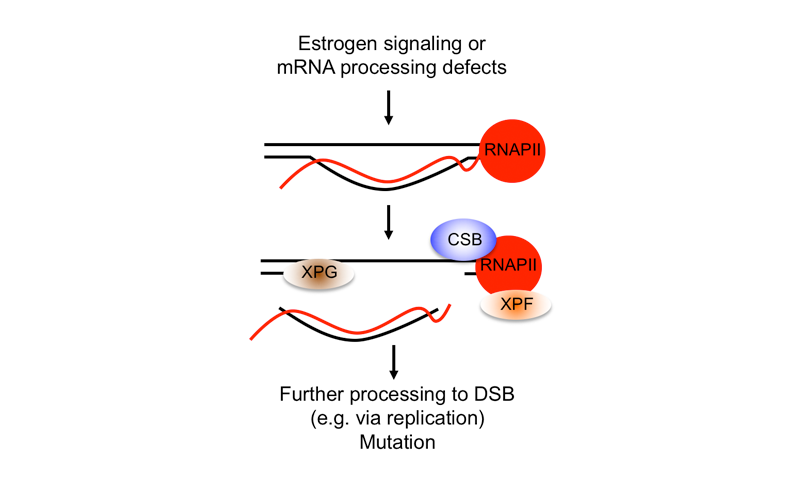
Several years ago, we carried out a genome-wide siRNA that suggested problems with transcription and mRNA processing posed a much more serious threat to genome stability in mammalian cells than we had previously appreciated. Further work showed that many transcription-associated factors and processes prevent DNA damage in mammalian cells by preventing the formation of R-loops, RNA-DNA hybrid containing structures which form when nascent mRNA reanneals with its DNA template. R-loops form naturally during transcription and have known regulatory roles in the cells, but they can also act as barriers to DNA replication, blocking progression of the replication fork and causing DNA damage and replication stress. Indeed, cells have evolved numerous pathways to regulate and resolve R-loops and to suppress their formation. Moreover, R-loop deregulation causes a variety of human diseases, including some neurodegenerative disorders, nucleotide expansion disorders and cancer.
We are interested in addressing several important questions about how R-loops induce DNA damage, specifically double-strand breaks (DSBs). Additionally, we are trying to understand how cells can tolerate regulatory R-loops and what causes an R-loop to become toxic. Importantly, we have discovered that R-loop-associated DSBs are a result of nuclease-mediated processing, and we have demonstrated that proteins involved in transcription-coupled nucleotide excision repair (TC-NER), a pathway known to repair bulky DNA lesions on the transcribed DNA stand, are needed to process R-loops into DSBs. In studying the role of R-loops in cancer associated genome-instability, we have also found that estrogen, a known mutagen and carcinogen in breast tissue, leads to S-phase and R-loop dependent DNA damage in breast epithelial cells. TC-NER factors also process these R-loops, which are induced predominantly in estrogen-responsive genes. Furthermore, we found that R-loops form in estrogen-responsive genes that are frequently mutated in breast cancers. This and other work suggests that R-loops are pervasive sources of genome instability in human cancers and that transcriptional changes associated with carcinogenesis due to changes in growth factor signaling, oncogene activation and other transcriptional regulatory processes can promote R-loop formation. We are now building on this initial work to study the molecular mechanisms involved in R-loop processing, and the relationship between R-loop formation and tissue-specific patterns of mutations in cancer. We are also exploring the potential to exploit R-loops in the treatment and diagnosis of cancer.
Please see the following publications to learn more about this work:
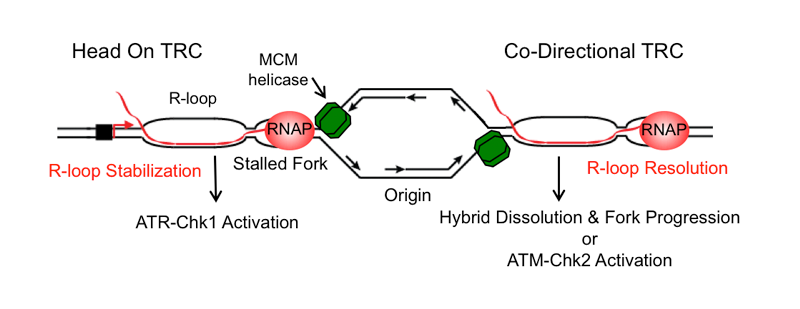
Transcription and replication are two fundamental processes that occur on the same DNA template, and the molecular machineries involved in both processes can come together in the form of transcription-replication ‘collisions’. If not properly controlled, such events can impede DNA replication leading to replication stress and DNA damage. Co-transcriptional R-loops may aggravate such collisions by stabilizing the transcription complex, creating an additional barrier to replication fork progression. Mechanistically studying transcription-replication collisions has been challenging because they can occur in two orientations in the genome (head-on/HO and co-directional/CD), and because the plasticity of eukaryotic replication programs allows different genomic loci to also undergo both types of collisions. Thus, we developed a novel episomal system to study transcription-replication collisions (TRCs) in human cells in a mechanistically tractable fashion, with a defined collision orientation at both R-loop forming and non-forming sequences. We have made some surprising observations with this system. For example, we have found that the direction of replication fork movement modulates R-loop formation and the outcome of TRCs, leading to distinct DNA damage responses.
Indeed, our work suggests that HO collisions promote R-loop formation while CD collisions resolve them. We validated this finding in the native genomic context using genomic approaches. Additional findings also suggest that proper execution of the replication program helps suppress TRCs and R-loop formation, providing mechanistic insight into the genome instability that arises in cancers where the replication program is deregulated. In future studies, we hope to further study the mechanisms involved in the resolution and cellular response to different types of TRCs, taking advantage of the versatile nature of this system and using other genomic and imaging approaches as well.
Please see the following publications to learn more about this work:
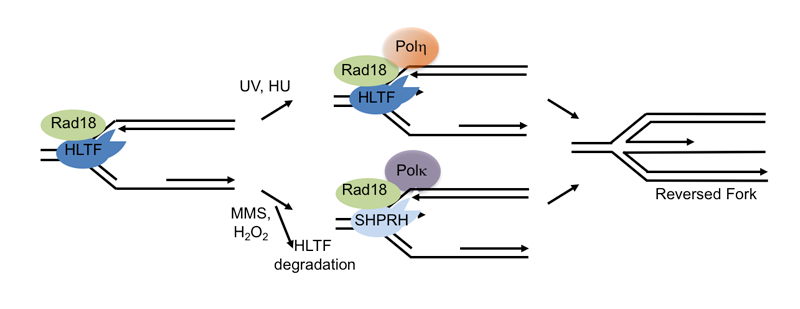
Replication stress is commonly observed in cancer cells and can lead to stalled replication forks, structures that can cause genome rearrangements if not stabilized or resolved. Cells use specialized translesion synthesis (TLS) polymerases to bypass some fork blocking lesions in an error-prone fashion, but they can also protect the stalled fork by remodeling it into a structure known as a reversed fork. Fork remodeling allows repair or resolution of the fork-blocking obstacle, promoting fork restart and continued DNA replication, but it can also allow template switching and lesion bypass using the sister chromatid. Bypass due to use of TLS polymerases or template switching is known as DNA damage tolerance (DDT). Intriguingly, there are numerous DNA translocases involved in these remodeling and DDT processes, including HLTF, SHPRH and SMARCAL1, all of which have the biochemical activity needed to remodel replication forks. Why cells need multiple enzymes with this activity is not clear and how they choose between error-prone and error-free pathways is poorly understood.
Our recent work has provided some important insights into the reasons for the apparent redundancy and pointed to how cells choose between different lesion bypass pathways, but it also raises many important questions. In addition to showing that two effectors of lesion bypass and fork reversal – HLTF and SHPRH – are regulated through their interactions and stability in a damage-specific manner, we showed that HLTF specifically remodels one type of replication fork structure. This work suggests that translocases use different mechanisms to recognize and remodel forks, and raises the possibility that these enzymes are specialized to remodel forks stalled by different obstacles or at different regions of the genome, questions we are seeking to address. Strikingly, we also showed that HLTF actively slows fork progression following replication stress in vivo, raising interesting questions about the consequence of a defect in this process. In future work, we hope to build on these and other results to understand why multiple fork remodelers exist, the mechanisms involved in regulating lesion bypass, fork remodeling and template switching, and the impact of their deregulation on mutagenesis. We are also seeking to understand the links between fork remodeling pathways, mutagenesis and cancer.
Please see the following publications to learn more about this work:
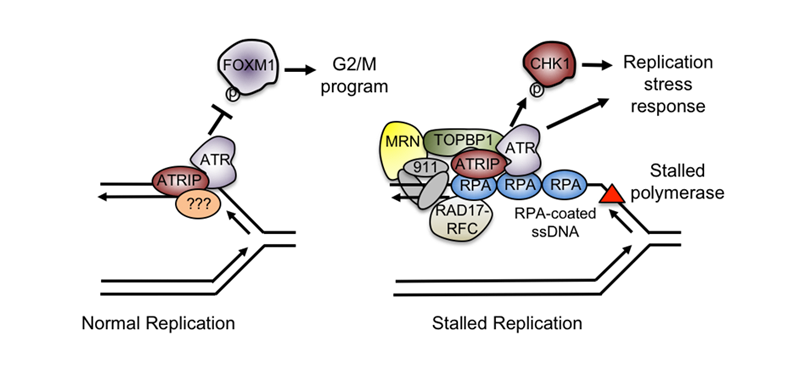
To efficiently and accurately complete DNA replication, cells must carefully coordinate replication fork progression with DNA damage responses and other cellular events. We are interested in understanding how DNA damage and replication stress responses are initiated, and how they regulate DNA replication, DNA repair and the cell cycle. In this regard, one focus of the lab has been on the replication stress response mediated by the ATR kinase, which I identified as a postdoctoral fellow. Using Xenopus egg extracts, the lab has studied the biochemical signal ATR senses at stalled forks, how replication and other processes trigger formation of this signal, and what other factors coordinate the ATR-mediated replication stress response. Collectively, our work has shown that ATR senses DNA structures that arise when the replicative helicase becomes uncoupled from DNA polymerase, rather than the absolute rate of DNA replication. This finding provided a mechanistic basis for DNA replication-dependent ATR activation by a broad range of lesions and events that threaten genome stability.
We have also used simple, purified DNA structures to ask what structural elements are necessary and sufficient for ATR activation. With this system, we have also studied proteins implicated in ATR activation, showing that some modulate the formation of the DNA signal while others play more direct roles by recognizing this signal or recruiting ATR activators. These findings defined the minimal signal capable of triggering ATR activation and were crucial for understanding ATR activation in other contexts, such as at the telomere or after nucleotide excision repair. Building on this work, we are now striving to understand ATR’s essential functions and the role that it plays in unperturbed cell cycles. Our recent application of high-content quantitative imaging has allowed us to demonstrate that ATR is activated in every S phase and that it has an intrinsic function required for proper cell cycle progression. Moving forward, we are seeking to understand the molecular basis of this form of ATR activation and cell cycle control. We are also interested in identifying other factors involved in ATR activation and signaling, and in sensitizing cells to ATR inhibitors. These studies will provide insight into how to target cancer cells exhibiting high levels of replication stress induced by oncogenes or other factors.
Please see the following publications to learn more about this work: